PREPARATIVE BIOCHEMISTRY, 17(4), 423-433 (1987)
Jaroslav Flegr
Institute of Molecular Genetics Czechoslovak Academy of Sciences Flemingovo nam. 2, 16637 Prague 6, Czechoslovakia
A rapid and simple method for the isolation and purification of dsRNA is presented. The crucial step of this method is the extraction of proteins and DNA with acid phenol. After the extraction, only RNA is left in the aqueous phase. ssRNA contamination of the RNA preparation can be greatly reduced when ammonium sulfate is present during the extraction.
INTRODUCTION
Double-stranded RNA can be isolated from cells and tissues by several different methods. Most often, total nucleic acid extracts from cells or from dsRNA-enriched fractions of cell homogenates are fractionated either by chromatography [1] or by selective precipitation with 4 M LiCl [2]. Both methods are time-consuming and often result in large losses of material.
We developed a one-step method to isolate and purify dsRNA from the protozoon Trichomonas vaginalis. DNA and proteins are extracted from the cells with phenol pH 4.0, which is followed by precipitation of dsRNA from the aquaeous phase with isopropanol. The method is rapid, inexpensive and gives very good yields of dsRNA. We showed that this approach was also suitable for the isolation of dsRNA from nonprotozoal material.
METHODS
Organisms.
Trichomonas vaginalis strain A1 [3] and Saccharomyces cerevisiae strain T i58 C (alpha, his -) (R.G.E. Palfree, Mc Gill University, Montreal, Canada) were used.
Isolation of dsRNA with phenol pH 4.0.
All steps of isolation were performed at room temperature. Trichomonads in growth medium ta tryptose, yeast extract, maltose medium (TYM) [4] with or without agarose) or in 0.8' NaCl (5-400 million cells/ml) were transferred to a plastic centrifuge tube, i volume of phenol pH 4.0 water-saturated phenol equilibrated with 50 mM acetate buffer pH 4.C) was added, and the tube was ' tightly capped and vigorously;. shaken for 3 minutes. The phenol and aqueous phases were separated by centrifugation and the agueous phase was reextracted with 8 volumes of chloroform-isoamyl alcohol t4:11. After centrifugation the upper phase was transferred to a new tube, a ).75 volume of isopropanol was added, and nucleic acids were precipitated from the mixture by three cycles of freeing (in dry ice or liquid nitrogen) and thawing,. The precipitated RNA was collected by centrifugation, the pellet was washed with 60% ethanol, dried and dissolved in TE buffer 10 mM Tris, 1 mM EDTA, pH 8.0).
Isolation of DNA and RNA with phenol pH 8.0,
The same method as described above for dsRNA was employed, but phenol pH 8.0 (water-saturated phenol equilibrated with at first 1 M and then 0.05 M Tris-HCl pH 8.0)) instead of phenol pH 4.0 ) was used for extraction.
Isolation of DNA and RNA with chloroform.
Trichomonads in 0.8% NaCl (5-100 million cells/ml ) were lysed with guanidine hydrochloride or guanidine iscthiocyanate (final concentration 4 M, stock solutions 8 M and 6 M, respectively). The extraction was performed with two:.volumes of chloroform-isoamyl alcohol (24:lI 5 times and nucleic aciids were precipitated as described above.
Electrophoresis.
Tris-borate agarose gel electrophoresis C57 was used for nucleic acids separation (0.8% agarose, submerged minigels 7 x 8 cm, 3 V/cm, 3 hours). Nucleic acids were detected with ethidiue bronide, ng/ml of gel.
RESULTS AND DISCUSSION
As revealed by electrophoretic patterns of nucleic acids extracted from equal numbers of T. vaginalis cells by three different methods (Fig. 1), extraction with acid phenol gave the highest yields of dsRNA without any apparent DNA contamination.
Solubility of the nucleic acid pellet was also studied. TE buffer was added to the pellets and samples were taken sequentially in the course of i20 minutes as specified in the legend to Fig. 2. At the end of the experiment the samples were electrophoresed (Fig. 2). The dsRNA extracted by acid phenol dissolved completely in 30-60 minutes; dsRNA extracted with guanidine hydrochloride needed 120 minutes to dissolve. The guanidine isothiocyanate method gave a very poor yield of dsRNA.
Comparison of the electrophoretic patterns of dsRNA extracted from T. vaginalis with phenol (pH 4.4 as well as pH 8.0)
and with guanidine hydrochloride tor guanidine isothiocyanate) showed that one dsRNA zone was absent in the f ormer (Fig. 1). A similar effect was observed when
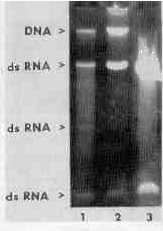
FIGURE 1
Nucleic acids of T. vaginalis isolated with guanidine isothiocyanate (lane 1), guanidine hydrochloride (lane 2), and acid phenol (lane 3).
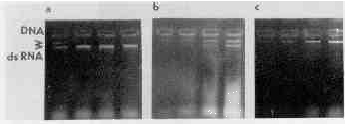
FIGURE 2
Dissolution of nucleic acids pellets. Pellets of nucleic acids isolated from 0 million trichomonads with acid phenol Ia), guanidine hydrochloride (b, or guanidine isothiocyanate tc were dissolved in TE buffer for 15, 30, 60, or 120 minutes. The arrows point to the ones of DNA (near the start of electrophoresis) and RNA.
dsRNA had been extracted from the yeast Saccharomyces cerevisiae. In this case the extraction was started from SDS-lysed protoplasts (prepared according to Eddy and Williamson [6]). Figure 3 shows that the L species of dsRNA (molecular mass 3.2 megadaltons [7] was extracted with phenol far more effectively and the M species (molecular mass 1.0-1.3 megadaltons [8]) far less effectively than by the guanidine hydrochloride method. This phenomenon could have been due to the presence of proteins covalently bound to these dsRNAs, which had made them enter the phenolic phase or the protein interphase [9].

FIGURE 3
RNA of Saccharomyces cerevisiae extracted with acid phenol (lane 1 and guanidine hydrochloride (lane 2).
The acid phenol method of isolation of dsRNA resembled a hot phenol method for the isolation of ssRNA C147; both extraction procedures eliminate the DNA into the phenol phase, leaving the RNA in the aqueous phase. Our results showed that the acid phenol method could be used for isolation of ssDNA and appeared to be even more effective than the hot phenol method. which must be repeated several times to achieve 95% elimination of DNA [10,11]. The quality of the ssRNA obtained is under investigation now, because of the reported depurination of nucleic acids at low pH [12] and elimination and partial degradation of poly(A)RNA with phenol [11, 13]. We observed that when a certain amount of ammonium sulfate (not NaCl or KCl) was added to the extraction mixture (Fig. 4), most of the ssRNA could enter the phenol phase. (For details see the legend to the figure). When high concentrations of ammonium sulfate were used, the water phase turned denser than the phenol phase and accumulated at the bottom of the centrifuge tube. Moreover, during isopropanol precipitation of dsRNA two distinct phases. an isopropanol and an ammonium sulfate-saturated water phase, appeared. In that case ammonium sulfate had to be extracted from the lower aquaeous phase with 60% ethanol before a pellet of RNA could be obtained by centrifugation.
Phenol extraction usually removes proteins, leaving DNA and RNA in the aqueous phase. It has been demonstrated, however, that under conditions of a particular pH, ionic strength or
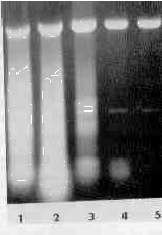
FIGURE 4
Elimination of ssRNA contaminant by ammonium sulfate. Before the extraction the samples (aliquots of trichomonads in 0.8% NaCl) were saturated to 20, 30, 40, 50 and 60% with ammonium sulfate (solid) (lanes 1-5, respectively).
temperature, it removes DNA and some classes of RNA with the proteins. Several methods of isolation of total ribonucleic acids [14], mRNA [11, 15, 16], rRNA [10] or covalently closed circular DNA (cccDNA) [17] are based on this phenomenon, the physical basis of which is not fully understood. In many systems a coprecipitation of DNA or poly(A)RNA with denatured proteins or with crystals of potasium dodecylsulphate is considered to be the most probable explanation [10, 11]. In the case of our method, as well as that of Zasloff et al. [17] (isolation of covalently closed circular DNA), however, a different mechanism must be operating, because DNA can be selectively extracted into the phenol phase from protein-free solutions. Both methods share the acid phenol extraction step. Zasloff et al [17] demonstrated that when phenol pH 4.5 instead of pH 4.0 was used, the DNA stayed in the aqueous phase. The methods differ, however, in their sensitivity to variation of ionic strength. When Zasloff et al extracted NA from buffers containing more than 200 mM NaCl, all nucleic acids entered the phenol phase (their material did not contain dsRNA). Our results showed that isolation of dsRNA is insensitive to an extensive variation of the concentration of ammonium sulfate (Fig. 4), NaCl or KCl.
From a practical point of view, the most important difference between our and Zasloff's method is that dsRNA can be isolated directly from cell suspensions while the isolation of covalently closed circular DNA requires previous elimination of proteins by extraction with phenol pH 8. It follows that using Zasloff's method, dsRNA, if present, could contaminate cccDNA extracts, while when isolating dsRNA by our method, no contamination of dsRNA extracts by cccDNA can be expected.
ACKNOWLEDGMENT
I thank Dr. J.Čerkasov (Charles University, Prague) for his advice and aid and Drs. J.Kulda (Charles Univereity) and M.Muller (Rockefeller University, New York for reading and commenting on the manuscript.
REFERENCES
1. R.M. Franklin, Proc. Natl. Acad. Sci. U.S.G., 55, 1504-1511 (1966).
2. J.R. Dia-Ruiz and J.M. Kaper, Prep. Biochem., 8. 1-17 (1978).
3. J.G. Meingassner and J. Thurner, Antimicrob. Agents Chemother., 15, 254-257 (1979).
4. L.S Diamond, J. Parasitol., 43, 488-490 (1957).
5. T. Maniatis, E.F. Fritsch, and J. Sambrook,"A Laboratory Manual of Molecular Cloning", Cold Spring Harbor, New York, (1982).
6. A.A. Eddy and D.H. Williamson, Nature, 179, 1252-1253 (1953).
7. D.J. Tipper and K.A. Bostian, Microbiol. Rev. 48, 125-156 (1984).
8. J.D. Welsh and M.J. Leibowitz, Nucl. Acids Res., 8, 2365-2375 (1980).
9. F. Hishinuma, K. Nakamura, K. Harai, R. Nishisawa, N. Gurge, and T. Maeda, Nucl. Acids Res., 12, 7581-7597 (1984).
10. K.S. Kirby, Methods in Enzymology, 12B, 87-99 (1968).
11. G. Brewerman, J. Mendecki, and S. Y. Lee, Biochemistry, 11,637-641 (l972).
12. T. Lindahl and B. Nyberg, Biochemistry, 11, 3614-3618 (l972).
13. R.P. Perry, J. L. Torre, D.E. Yelley, and J.R. Greenberg, Biochim. Biophys. Acta, 262, 220-226 (1972).
14. J.R. Warner, R. Soerro, H.C. Birnboin, M. Girard, and J.E. Dornell, J. Molec. Biol., 19, 349-361 (1966).
15. P.M. Liardi, Methods in Enzymology, 96J, 24-38 (1983).
16. G. Brewerman, Biochim. Biophys. Acta, 76, 322-324 (l963).
17. M. Zasloff, G.D. Ginder and G. Felsfeld, Nucl. Acids Res.,5, l139-1152 (1978).