Praktikum k přednášce Molekulární ekologie
Populační struktura a sekvence
DNA
Rozdělení sekvencí do haplotypů, haplotypová síť, FaBox, TCS
Úloha*:
V programu Geneland zjistěte optimální K a zařaďte jedince dle jejich genotypů do skupin.
Zkuste i prostorový model.
Geneland pracuje v prostředí R. Nejprve spustíme R a musíme nalézt a nainstalovat balík (Package) Geneland. Stáhneme ho odtud:
https://i-pri.org/special/Biostatistics/Software/Geneland/
Geneland spustíme příkazy:
library(Geneland)
Geneland.GUI()
Otevře se příjemné grafické prostředí, ve kterém je již analýza poměrně snadná.
Šikovné je zadat Thinning. Určuje, kolik kroků analýzy bude zaznamenáno. Např. když zadáme 100, bude zaznamenán každý stý krok, což je většinou dostačující.
genotypy
souřadnice
Úloha*:
Alignment sekvencí "ořízněte" na délku nejkratší sekvence. Sekvence rozdělte do haplotypů. Zjistěte zastoupení jednotlivých haplotypů. alignment
Postup: Můžeme použít programový balík FaBox http://users-birc.au.dk/palle/php/fabox/ Funguje online a hodí se na spoustu šikovných úprav souborů ve FASTA formátu. My využijeme možnost Alignment cropper (pro oříznutí sekvencí) a DNA to haplotype collapser and converter (pro rozdělení do haplotypů).
Doporučené další možnosti:
V programu FaBox vyzkoušejte editaci názvů sekvencí a práci s alignmenty. Při práci se sekvenci vám znalost těchto možností ušetří spoustu času a chyb.
Haplotypovou síť můžeme vytvořit v programu TCS Vstupní soubor vytvoříme lehce z "ořezaného" alignmentu opět ve FaBoxu (možnost Create TCS input file).
Jsou ale i šikovnější možnosti:
Úloha*:
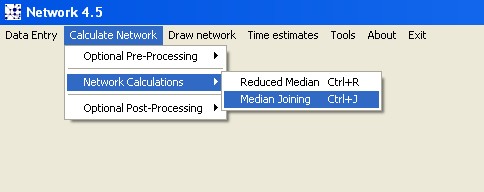
Sestrojte síť haplotypů v programu Network http://www.fluxus-engineering.com/sharenet.htm Vstupní soubor můžete použít stejný jako pro TCS. POZOR! Vstupní soubor poněkud nelogicky zadejte přes možnost "Calculate network" hlavního menu!
Úloha*:
Sestrojte síť haplotypů v programu PopArt
Pozor! Jiný formát vstupního souboru (Nexus)
http://popart.otago.ac.nz/downloads.shtml
Populační struktura a sekvence
DNA
Mismatch distribution
Úloha*:
Vypočtěte "number of segregating sites", "haplotype diversity" a "nucleotide diversity"
Postup: Můžeme vhodně použít program Arlequin, který si stáhneme ze stránky: http://cmpg.unibe.ch/software/arlequin35/Arl35Downloads.html
Data (soubor skupiny a populace) importujeme do programu (karta "Import data").
haplotypy
vzorová populace
Úloha*:
Odhadněte demografickou historii jedné populace pomocí "Mismatch distribution"
Postup: Opět můžeme použít Arlequin.
Data z předešlé úlohy:
haplotypy
vzorová populace
AMOVA (program Arlequin)
Úloha*:
Vypočtěte hierarchickou analýzu molekulární variance (AMOVA).
Jedinci jsou zařazeni do populací. Populace tvoří skupiny ("Groups").
Kolik procent variability nám vysvětlí rozdělení na "Groups", kolik vysvětlí rozdělení na populace v rámci groups?
Kolik procent variability nám pak zbyde na rozdíly v rámci jednotlivých populací?
Postup: Opět můžeme použít Arlequin.
Vstupní data:
haplotypy
Zde máme všechny haplotypy, které se vyskytují v našich populacích. Soubor je vhodné uložit do stejného adresáře jako vstupní soubor:
skupiny a populace
Zde je popsáno, v jakých populacích se vyskytují konkrétní haplotypy a v jakém počtu. Na konci souboru je pak napsáno, jak populace rozčlenit do "Groups". Na kartě "Settings" pak zadáme možnost AMOVA.
Isolation with migration
Úloha*:
Odhadněte míru izolace, směr toku genů, dobu divergence a efektivní populační velkosti dvou částečně izolovaných populací.
Využijeme program IMa ze stránky:
https://bio.cst.temple.edu/~hey/software#im-div
Vstupní soubor zde
Příklad programového řádku pro spuštění programu:
ima -i inputPavel.txt -o output.txt -q1 10 -m1 10 -m2 10 -t 10 -b 1000000 -L100000 -p 45 (jeden řetězec)
ima -i inputPavel.txt -o output1.txt -q1 10 -q2 200 -m1 1 -m2 1 -t 10 -b 1000000 -L100000 -p 45 -n 10 -f g -g1 (více řetězců)
Výstupní soubor zde
IM2
Příklad programového řádku:
IMa2 -i mtInput.txt -o trialrun.out -q 80 -m 8 -t 5 -b 100000 -l 50000 -hfg -hn 20 -ha 0.96 -hb 0.9 -d 20
Vzor vstupního souboru pro sekvence a pro mikrosatelity