
Structure prediction
|
|
Bioinformatics:
From Genome Sequences to Protein Structures Structure prediction |
| Page 5 of 5 |
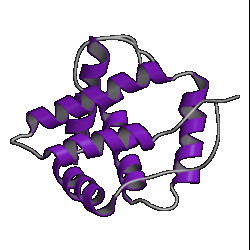
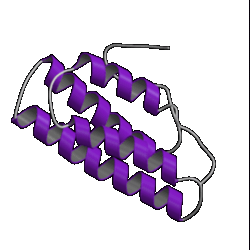
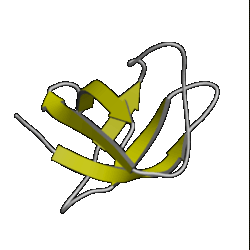
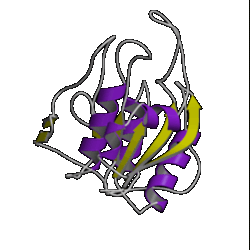
There are currently over 670 unique folds in the PDB. Some estimates put the number of protein folds found in Nature at around 1,000. Some folds occur more often than others, such as the 10 superfolds shown below.
|
|

|

|
|---|---|---|---|
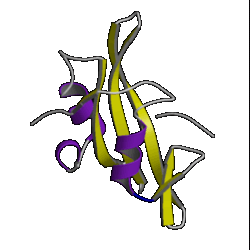
| 1. globin ( 1hlm ) |
2. updown ( 2hmz ) |
3. trefoil ( 1i1b ) |
4. Tim barrel ( 7tim ) |
|
|

|
|
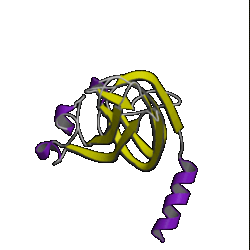
| 5. OB roll ( 1mjc ) |
6. doubly wound ( 3eca ) |
7. immuno- globulin ( 3hhr ) |
8. UB roll ( 1sha ) |
|
|
||
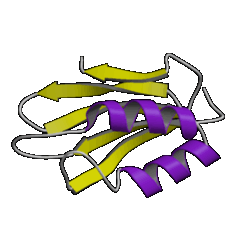
| 9. jelly roll ( 2stv ) |
10. plaitfold ( 1aps ) |
||
Current methods of fold recognition are not particularly reliable. They are most likely to fail if the query sequence corresponds to a multi-domain protein, or if its fold is not one of those in the PDB.
There are a number of protein fold recognition servers on the Web:-
All these methods can take a very long time to run. So, here is a set of results prepared earlier. The results were generated by submitting the sequence to the Phyre Web server.
Simply scroll down the upper box to look at the results. The lower box contains the probe sequence, a secondary structure prediction and its assigned reliability. Briefly compare the prediction with the results from the Jpred consensus secondary structure prediction.
In the table in the upper frame the significance of each hit is given by confidence scores:-
The top hit in the table, is, happily, the true structure (1sro) whose fold is an OB-fold - a type of mainly-beta barrel. The next 7 hits are also OB-folds, which is encouraging, but remember that these results have been obtained from a database containing the structure we're looking for, so are likely to be biased towards giving a correct answer. Such an apparently high level of success is not always obtained when the sequence is genuinely of a protein whose structure is unknown.
At the time our sequence was being used as Target 4 for the Casp2 fold recognition experiment, the 1sro structure had, of course, not been published. Of the 708 fold predictions made for this sequence the following PDB codes were most frequently given as being compatible with the target sequence:-
| PDB code |
Protein | Fold | Number of times matched |
|---|---|---|---|
| 1csp | Major cold shock protein | Beta-barrel | 33 |
| 1mjc | Major cold shock protein | Beta-barrel | 27 |
| 1bov(A) | Verotoxin-1 | Beta-barrel | 7 |
|
1lyl(A)
(dom 1) |
Lysyl-tRNA synthetase | Beta-barrel | 7 |
Which of these are also in the Phyre hits?
Let's see how these folds compare with the correct answer (ie
the structure of 1sro). We will use the  database to align and superpose the above proteins so that we can view
them in 3D using RasMol.
database to align and superpose the above proteins so that we can view
them in 3D using RasMol.
The steps are as follows:-
AEIEVGRVYTGKVTRIVDFGAFVAIGGGKEGLVHISQIADKRVEKVTDYL |
So, type the following series of PDB codes and chain identifiers into the lower of the two boxes. (In fact, you can just cut-and-paste from here!):-
The sequence identities in the
alignment are given at the bottom of the lower frame. As you might
expect the mystery sequence is 100% identical to 1sro.
Its identities with the other sequences are all in the 20-35%
Twilight Zone. (The one apparent exception of 1bov(A), which has
a sequence identity of 43.5%, is not significant as it only applies
over a short segment of 23 residues).
In the new version of the alignment the blue residues represent residues in beta strands, while the red ones represent residues in helices.
Comparing all the sequences suggests that the patterns of secondary structure regions match quite well.
Compare the structures in SwisPDBviewer.
However, don't be lulled into a false sense of confidence in these methods. This was a special case which was relatively easy to predict. Have a look at the picture of the 10 superfolds given at the top of this page. You'll find 1mjc is one of them! In other words, our mystery sequence adopts one of the superfolds. The structure is a small one and the database contains many examples of this fold (although none are above the Twilight Zone in terms of sequence similarity to our mystery sequence). So it should be a relatively easy sequence to predict.
In all, CASP2 had 15 targets for the fold recognition exercise, of which our mystery sequence was one. These were classified as:-
where the classification from easy to medium to hard was based on the nature and extent of the solved structure's similarity to existing structures in the PDB. The impossible structures were those with no recognizable similarity to anything in the PDB .
Our mystery sequence was one of the two easy targets. And these were indeed fairly easy; a number of methods succeeded in identifying the folds correctly. The prediction success declined with increasing difficulty of the targets, and the impossible targets were not always recognised as being impossible.
 |
This material is prepared with the support of the project ESF pro V� II na UK, Reg. num.: CZ.02.2.69/0.0/0.0/18_056/0013322.