
Structure prediction
|
|
Bioinformatics:
From Genome Sequences to Protein Structures Structure prediction |
| Page 1 of 5 |
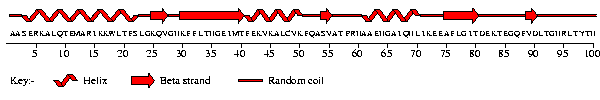
The predictions can be rather inaccurate, so be warned. The best methods currently achieve around 70% accuracy.
However, a prediction may at least be useful in suggesting which of the four fold classes, shown below, the protein falls into depending on its overall secondary structure content.
| Class 1 | Class 2 | Class 3 | Class 4 |
|---|---|---|---|

|

|
|

|
| Mainly alpha | Mainly beta | Alpha/beta | Few secondary structures |
This can narrow down the range of possibilities for, say, the fold recognition methods which we'll meet later in the tutorial.
AEIEVGRVYTGKVTRIVDFGAFVAIGGGKEGLVHISQIADKRVEKVTDYL |
This sequence was one of the "target" sequences submitted to CASP2 . It represents the S1 motif of polyribonucleotide nucleotidyltransferase from E. coli. A new CASP experiment is held every second year and the results can be found at the Protein Structure Prediction Center. In the present tutorial, however, we will concentrate only on CASP2.
| CASP stands for "
Critical Assessment of Techniques
for Protein Structure
Prediction" and the "2" in CASP2 refers
to the second of the CASP meetings (to date there have been
six such meetings). These are international experiments in assessing
how good/bad all the different structure prediction methods are.
The experiments involve releasing the sequences of proteins whose structures are currently being solved by crystallography or NMR groups around the world. Predictions are then invited in various categories. Once the structures have been solved the accuracy of the predictions can be assessed, and the results are presented at the end of each year at a specially convened conference. The above sequence was submitted to the fold recognition and ab initio categories of CASP2, and its 3D structure has now been deposited in the PDB. At the time, however, there was no close homologue of known 3D structure. |
The GOR prediction can be run interactively from the Network Protein Sequence @nalysis site.
What percentages of the different secondary structures are predicted?
| %-tage helix; | %-tage strand; | %-tage coil |
|---|
On the basis of this prediction, in which of the above fold classes would you place the protein?
In fact, the above prediction is actually only 48.7% correct - and gives the wrong fold class! But normally we wouldn't know that(!).
Go back to the NPS@ home page and click on the Predator method. Repeat the above steps to get the Predator predictions and fill in the answers below.
What percentages of the different secondary structures are predicted?
| %-tage helix; | %-tage strand; | %-tage coil |
|---|
On the basis of this prediction, which of the above fold classes would you place the protein?
Read the "Abstract" on the results page to see how the Predator methods differs from GOR.
This time the prediction is 52.6% correct. Barely more that half the protein! You may be wondering what use this is, but these are very simple methods, and accordingly suffer from a lack of accuracy.
Carry on HERE
 |
This material is prepared with the support of the project ESF pro V� II na UK, Reg. num.: CZ.02.2.69/0.0/0.0/18_056/0013322.