
Structure prediction
|
|
Bioinformatics:
From Genome Sequences to Protein Structures Structure prediction |
| Page 2 of 5 |
Accuracy can be improved by:-
|
|---|
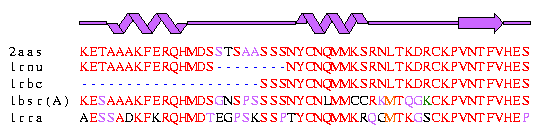
| Alignment of five ribonuclease A sequences. Each residue is coloured according to its similarity to the corresponding residues in the top sequence. The two helices and one strand are in regions which show the greatest degree of conservation (predominantly red) whereas the loops are associated with the regions of greater residue variability and of residue insertions/deletions. |
Sequence : AGACSTCAGKLVSGPAPDEDQSFLDDDQIQAGYILTCVAYP Method 1 : -----EEEEEEEE-------------------EEEEEE---
Method 2 : ---------EEE-----------H-----HHHEEEEEEE--
Method 3 : --------------------------------EEEEEE---
Method 4 : --------EEEE--------------HHH--EEEEEEEE--
Method 5 : -------EEEE---------------HHHHHHHEEEEE---
Method 6 : ---EEEEEEEEEEEE----------HHHHHH--EEEEEE--
Consensus : --------EEEE--------------HHH---EEEEEE---
Here the bottom line gives a prediction based on some sort of weighted consensus among the six methods.
One site which makes use of both the above strategies is:
Try to run it, but do not use information from available structures - that would make it too easy.
Which fold class does the above prediction suggest?
How much of the protein is predicted as helix?
Now have a look at the line labelled "JNET Rel" in FULL HTML results. This gives relative level of confidence in its prediction. The green regions (associated with scores of 7-9 ) represent the highly-confident regions, most likely to have been correctly predicted.
How many regions of regular secondary structure might you be reasonably sure are correct?
Now let's see what we have if we take only the highly-confident regions. Paste the consensus prediction into the box below. Then paste the " PHD Rel" column below it. Then, below that, retype the consensus prediction so that it consists of dots, ".", at residue positions of low confidence, and the predicted secondary structure at positions of high confidence.
For example:-
------EEEEEEEEEE-Confidently predicted regions
97242258677767653
--.....E.EEE.E...
What percentage of the sequence has been confidently predicted?
Of particular interest are alpha-helices or beta-strands which are either amphipathic or totally buried.
|
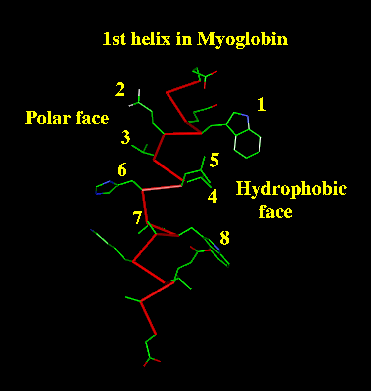
An amphipatic helix is one which, because it is on the
surface of the protein, has one side consisting largely of hydrophobic
residues which face the protein's hydrophobic core, and the opposite
side consisting largely of polar residues which face out into the solvent.
As alpha-helices have a periodicity of 3.6 residues per turn, the pattern of buried (B) residues will be of the form: i, i+3, i+4, i+7. Such a pattern would suggest a surface helix (as in the example shown, where i=1). |
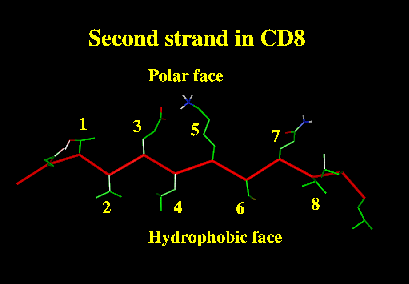
| Similarly, an amphipatic strand has one side hydrophobic
and the other polar. The geometry of strand residues means that
the pattern of buried (B) and exposed (E) residues
is a simple alternating one.
In this case the pattern suggests the strand is the "edge" strand of a beta-sheet, and pokes out into the solvent. |
|
|
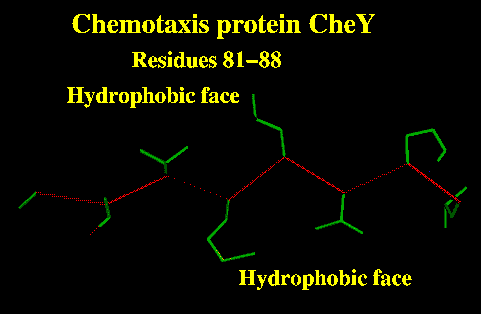
Beta-strands in alpha/beta proteins are often completely buried.
These can be identified by a run of hydrophobic residues.
In the Jpred prediction this would show up as a run of residues classified as buried (B). |
Can you identify any of the above features in the Jpred accessibility predictions for our mystery protein?
Carry on HERE
 |
This material is prepared with the support of the project ESF pro V� II na UK, Reg. num.: CZ.02.2.69/0.0/0.0/18_056/0013322.