
Structure prediction
|
|
Bioinformatics:
From Genome Sequences to Protein Structures Structure prediction |
| Page 3 of 5 |
Now let's see how well the methods did. After all, we know that the structure of the protein has in fact been solved and is now in the PDB. Its PDB code is 1SRO . You may download the PDB file by clicking the right mousebutton here (and choose "save target as" or "save link as" - depending on the browser you use).
Here's its "correct" secondary structure, as calculated using the DSSP algorithm:-
1 2 3 4 5 6 7
1234567890123456789012345678901234567890123456789012345678901234567890123456
.........:.........:.........:.........:.........:.........:.........:......
AEIEVGRVYTGKVTRIVDFGAFVAIGGGKEGLVHISQIADKRVEKVTDYLQMGQEVPVKVLEVDRQGRIRLSIKEA -------EEEEEEEEEE--EEEEE---------------------HHHH-----EEEEEEEE-------EEEE---
The first thing to note is that the sequence above is 8 residues shorter than the sequence we were given to predict. Presumably the experimental data were insufficient to uniquely define the structure of the C-terminal end of the protein. We will merely ignore the last 8 residues from our original sequence when assessing the accuracy of the prediction methods.
What percentage of residues are in the "core" regions of the Ramachandran plot? What does this tell you about the quality of the model?
(If you're not sure how to get this information, go to the PDBsum page for 1sro (PDBsum) and click on the PROCHECK summary button).
In fact, rather than giving a single model of the protein's structure, the 1sro entry contains an ensemble of 20 slightly different models (not untypical for structures solved by NMR) and, within the ensemble, there is some disagreement as to the secondary structure of the protein(!). For example, here are the first 50 residues of models 1 and 2 in the ensemble:-
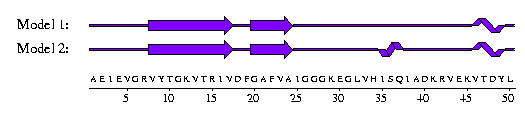
The second model has a small helix at residues 35-37, not present in the first. This simply means that the hydrogen-bonding patterns that DSSP recognises as characteristic of alpha-helices are present in Model 2 but not in Model 1.
What's more, if you look closely at the structure in your favourite molecule viewer (e.g. RasMol or SwissPDBviewer) , you will suspect that maybe there should be another strand in the structure which may have been missed by the DSSP algorithm. There is a loop which looks like it should be part of a beta-sheet yet presumably lacks the appropriate H-bonding patterns. This could be due to the relatively poor quality of the NMR model. (In related X-ray structures, this loop is indeed a strand in a beta-sheet).
Which range of residues looks like it might belong to a "missed" strand?
There are two "take-home" messages here:-
Firstly, look again at the PDBsum page for 1sro and note down the protein's actual fold (as given by its CATH classification).
What is the protein's fold type?
Now for the secondary structure predictions. Below are the predictions you will have obtained from the GOR IV and PREDATOR methods. Paste them into the box below.
Sequence: AEIEVGRVYTGKVTRIVDFGAFVAIGGGKEGLVHISQIADKRVEKVTDYLQMGQEVPVKVLEVDRQGRIRLSIKEA
GOR IV: ----EEEE----EEEEE----EEEE-------HHHHHHHHHHHHHHHHHHHH-----EEEEEE----HHHHHHHHH
PREDATOR: ---EEEEE----EEEEEE---EEE-------EEE-----HHHHHHH------------EEEE-----HHHHHHHHH
Now add the Jpred consensus prediction and the "correct" secondary structure from above.
If you have time (and energy) see how well the regions predicted with "high-confidence" by Jpred fare.
How well do you think the predictions have done overall? How useful are they? Any other observations?
In fact, as we will see later, our mystery sequence is actually quite an "easy" target, and the prediction accuracy higher than average. The prediction methods usually perform worse than this.
Carry on HERE
 |
This material is prepared with the support of the project ESF pro V� II na UK, Reg. num.: CZ.02.2.69/0.0/0.0/18_056/0013322.