
Structure prediction
|
|
Bioinformatics:
From Genome Sequences to Protein Structures Structure prediction |
| Page 4 of 5 |
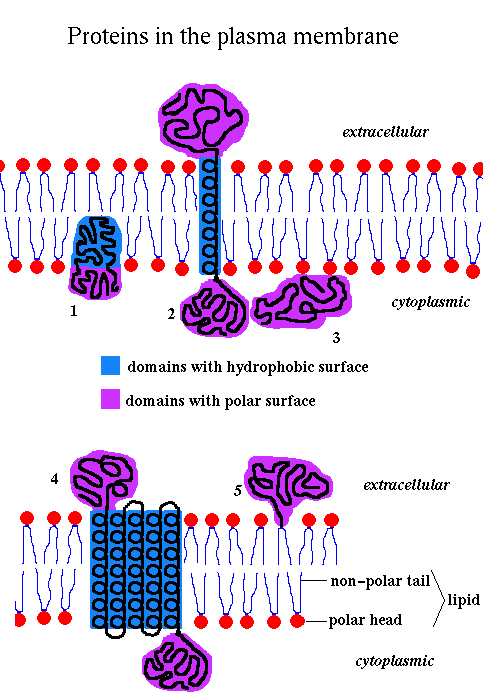
There are several types, here numbered 1-5:-
|
|
|---|
In all the above examples, the regions of the protein that are within the membrane, and which interact with the membrane's lipid bilayer, have to primarily consist of hydrophobic residues due to the bilayer's hydrophobic character. The regions outside the membrane, which are exposed to the aqueous solution, have a surface polarity comparable to that of soluble proteins. The interior of a membrane protein can also be very polar as is the case, for example, for channel proteins.
Here we are concerned with proteins of type 4 - those with membrane spanning regions which have a characteristic pattern of hydrophobic and polar regions.
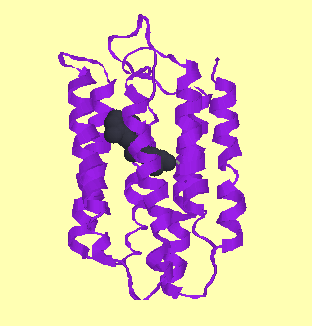
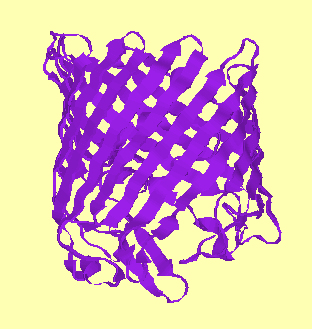
They fall into two categories: alpha-helical bundles and beta-barrels:-
| Bacteriorhodopsin (alpha-helical bundle) |
Matrix porin (beta-barrel) |
|---|---|
|
|
| PDB code: 1ap9 | PDB code: 1opf |
To span the membrane, the alpha-helices need to be around 20 residues in length, and the beta strands around 12.
The polypeptide chain alternately passes from the outside of the cell to the inside and then back again:-
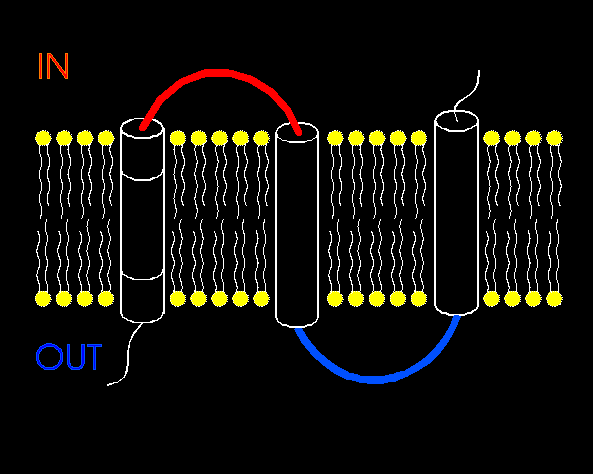
Methods which try to predict the transmembrane-spanning regions of a given protein sequence rely on identifying these characteristic patterns of hydrophobic and polar residues, specifically in transmembrane helices. Given a sequence that is known to be that of a membrane protein, the prediction of transmembrane helices can reach accuracies of 95% (Rost et al., 1995 ), while the fraction of false positives (ie globular proteins predicted as having one or more transmembrane helices) is about 2% (Rost et al., 1996 ).
Let's try to see what one such prediction method gives for our mystery protein. Here's the sequence again:-
AEIEVGRVYTGKVTRIVDFGAFVAIGGGKEGLVHISQIADKRVEKVTDYL |
Paste the sequence into the large box and click on the "Run TMpred " button.
This will give you a list of possible transmembrane helices and a model of how the protein might sit in the membrane.
Are there any TM helices?
Number of helices?
Now compare the prediction with the secondary structure predictions from Jpred.
Do the predictions agree or conflict?
Let's have a look at the TMpred prediction for a genuine membrane protein. Run the prediction for the bacteriorhodopsin sequence:-
XXXXXXRPEWIWLALGTALMGLGTLYFLVKGMGVSDPDAKKFYAITTLVPAIAFTMYLSM |
How many helices are given by the strongly preferred model?
Let's see how well the prediction did:
You should get something that looks like:
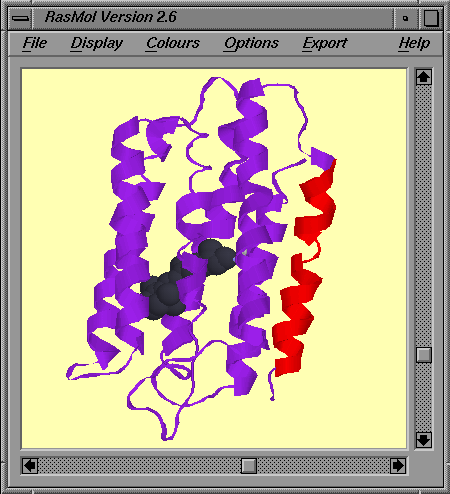
How well do you think the prediction has done?
Compare the scores given for each of the predicted transmembrane helices with those we got for our mystery protein sequence above.
Carry on HERE.
 |
This material is prepared with the support of the project ESF pro V� II na UK, Reg. num.: CZ.02.2.69/0.0/0.0/18_056/0013322.