 Part 2: Sequence Alignment
Part 2: Sequence Alignment
For this part you will use several of the softwares listed below, make sure you have them.
Programs
Aligners:
Alignment trimming:
Tree viewing and manipulation:
Alignment of nucleotide and amino acid sequences
Command line method:
To see MAFFT options type in the following command:
mafft -h
By default, MAFFT will determine the best algorithm based on the number of sequences. However, for most applications, L-INS-i shows the best performance. Thus, the basic usage would be:
mafft --localpair --maxiterate 1000 INFILE > OUTFILE
Open the resulting alignment with AliView. If the alignment has too many gaps, try increasing the gap opening penalty by setting it to 2-5 (compare with gap extension penalty):
mafft --localpair --maxiterate 1000 --op 3 INFILE > OUTFILE
What is the output file format?
Exercise 1
1. Download the sequences of Heat shock protein 90.
You can right-click and select
Save Link As...to download the file.
2. Use MAFFT to align the sequences. Remember to replace PLACEHOLDERS INFILE and OUTFILE with actual file names! Use the local mafft first, or if experiencing problems, go ahead with the online version below.
After you paste or type
mafft(with any desired parameters), start typing the input file name and complete it withTab.
Online method:
https://mafft.cbrc.jp/alignment/server/In the online method, we can input data either by pasting into the input field, or by uploading our FASTA file
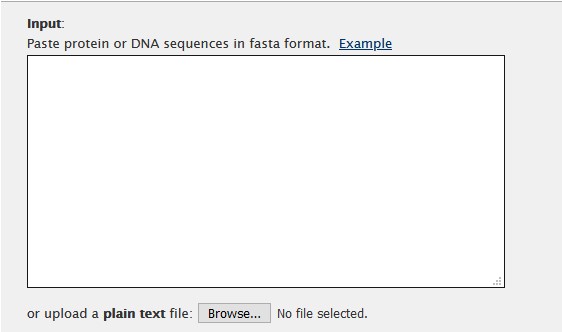
The algorithm can be selected in the Advanced settings section. For this example we will use L-INS-i algorithm.
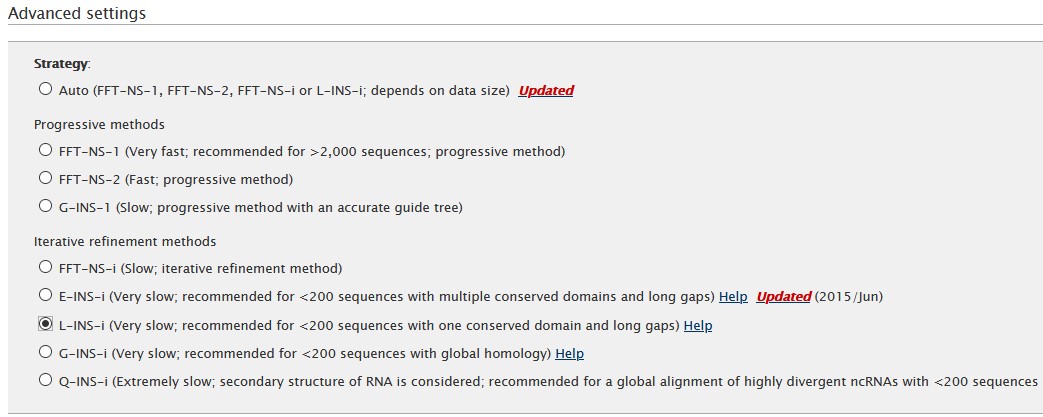
Finally, other parameters like the scoring matrix and gap opening penality, can be set in the Parameters section.
After setting all parameters we can hit the submit button. The server will compute the alignment and after the computation finished, we can analyze the alignment in the browser. To save the aligned sequences, we can use the top menu. Several options for saving are available, but we will use FASTA format.

Alignment trimming
After making an alignment, it is necessary to inspect and trim it to remove non-homologous sites.
- Remove leading and trailing misaligned regions.
- Remove sites that are suspicious of being non-homologous (usually recognized as regions with high variability of characters).
- Remove gappy regions (with more than 20 / 30 / 50 percent gaps).
There are programs for automated alignment trimming:
Visual inspection is highly recommended though!
Manual trimming can be done in Geneious (licensed) or AliView. Drag and drop the alignment file to the program window and check for regions to remove. Select them and right-click to Delete selected sequences and confirm (twice first time – to allow editing and then deleting sequences).
You can use
Ctrl(Cmd) + Delto delete the selected regions.
Save file as FASTA or try saving as “strict” PHYLIP. What is the difference between the formats? What should we keep in mind?
Hint: reopen this strict PHYLIP file with AliView.
For comparison, we are going to use automated trimming by trimAl. First, let's get some help how to use trimAl:
trimal -h
Try different trimming options and compare the results. A few examples are suggested below:
trimal -in INFILE -out OUTFILE -fasta -automated1
trimal -in INFILE -out OUTFILE -fasta -gt 0.7
trimal -in INFILE -out OUTFILE -fasta -gappyout
Automated1 is a heuristic method, -gt removes sites with >30% gaps, -gappyout automatically computes a trimming score based on gap distribution.
Those who cannot run trimal locally, use the Phylemon2 webserver:
http://phylemon2.bioinfo.cipf.es/
Similarly we will also try trimming with BMGE. Let's get some help how to use BMGE:
bmge -?
BMGE has two main parameters to control the trimming: -g which stands for gap rate and -h which stands for entropy.
A few examples for trimming with BMGE are suggested below:
bmge -i INFILE -of OUTFILE -t AA
bmge -i INFILE -of OUTFILE -t AA -g 0.3
bmge -i INFILE -of OUTFILE -t AA -h 0.4
-tis an important parameter in BMGE. It specifies what type of sequences you have for trimming. It can beAAfor proteins,DNAfor DNA/RNA sequences, andCODONfor codon alignments.
Alignment based on RNA secondary structure
For this we will use the SILVA database.

From the SILVA database, one can download secondary-structure alignments of ribosomal RNA from selected taxa. For this use the Browser section from the top menu on SILVA.
If you are not certain about a taxon's lineage, you can search for it using the Search tab. Use a Latin species name from that taxon, then hit the rightmost icon to show the sequence in the Browser with its full lineage.
Once you have selected the sequences which you want to download, you just add them to the cart and then you can download the sequences directly from the cart menu located on the top right.
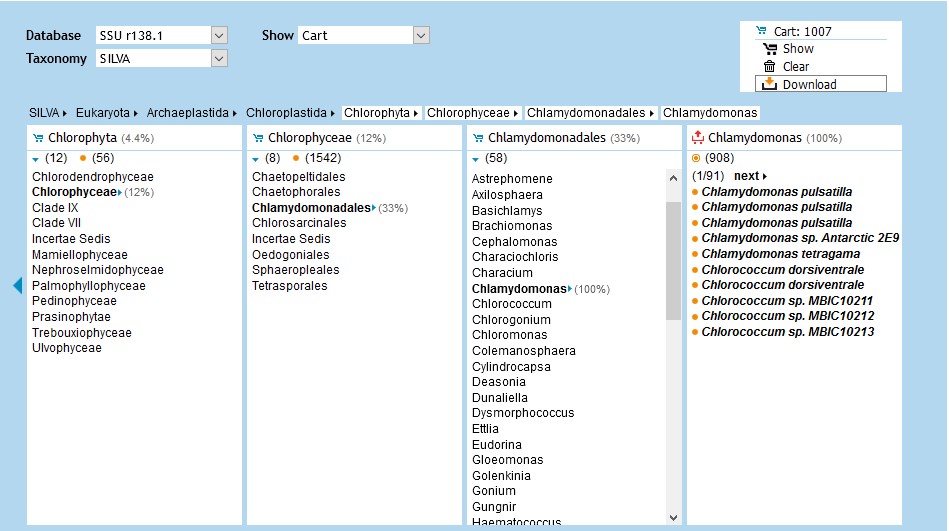
You can also align any sequence of interest using the Aligner found under the ACT tab.
Exercise 2
1. Download the mammalian SSU dataset from SILVA, using the browser function.
2. Using the ACT > SILVA Aligner, find the structural homology of the Northern treeshrew (Tupaia belangeri; tana severní) SSU.
>embl|AAPY01019204 Tupaia belangeri
ccctctattgttgtaatgtatagatagtccatatattaactgtgtctttatttaacaagatttctatctaggtctttgtactcagcaaagttatcaaactggcctgggactaaaactatcaaacatcctgcaaggcccctcttcaaaagacccttcccagtaatcaagatctaagaagatacacagacaaatgaacttcctcatcctgagaactgtgattgattttacctgatctgacaagacaaacagcatgttttataattattgcttccatactttgtataccctgttccttagagtaaattttttactacccctcaccctccttacttcctaaaccttcctcaataaataggcatgttttaatagccacggcacggcagcctggagctggttctggctgtggccagctgagaacatgctaaggagcaatgaggtctcagtctaatttttttgttcctgatcacccatgttgccttcctgtgaacgagccacacgccacggccgaggccagaccctggcattaagccgcaggctccacttctggtggtgcccttccgtcaattccttcaagtttcagctttgcaaccatactccccccagaacccaaagattttggtttcccagaagctgcccgatgggtcatgggaataatgctgccgcattgccagttggcatcatttatggtcggaactacaacggtatctgatcatcttcaaacctctgacttttattcttgattaatgaaaacattcttggcaaatgctttttctctggtccgtcttgtgcgggtccaagaattccacctctagcggcgcaatacaaatgcccccggccatccctcttaatcatggcctcagttccgaaaaccaacaaaatagaaccacggtcctattctattattcctagctgcggtatccaggtggcttggacctgctttgaacattctaattttttcaaagtaaatgctttgggccccgtgagacactcagctaagagcatcaaggggacaccgagaggcaaggggcgcctcgcagcagaccacccgcctgctcccaagatccaactacgagctttttaactgcagcaactttaatatacgctattggagctggaattaccatggctgctggcaccagacttgccctccaatagatcctagttaaaggatttaaagtggactcattccaattacagagcctcaaaagagtcctgtattgttattttttgtcactacctccccaggttgagagtgggtaatttgggcacctgctgccttccttggatgtggtagtcatttctcaggctccctctctggaatcgaaccccgattccctgtcatccatggtcaccatggtaggcacggcgactaccatcgaaagttgatagggcagacgtttgaatgggtcatcaccaccacagggggcgtgcgatcagcccgaggttatctagagtcaccaaagccaccggcacccgcacccccggcccaggtcgggggggagctgaccgttggttttgatctgataaatgcacgcatcccccccttgccgggggtcagcgcccgtcggcatgtattagctctagaattaccacagttatccaagtaggagaggagcgagcgaccaaaggaccataactgatttaatgagccattcgcagtttcactgtaccagccgtgggtacttagacatgcatggcttaatcttt
You can also find its neighbors, i.e. highly similar sequences from the SILVA database. Check the
Search and classifybox and setMin. identityto 0.5 and theNumber of neighboursto retrieve to 100.