 Part 1: Sequence Retrieval
Part 1: Sequence Retrieval
For this part you will use the software listed below.
Programs
Alignment editor:
Where and how to retrieve sequences?
Three of the most well known databases are GenBank, ENA and DDBJ. These three databases share a a large part of their data.
Other more specialised databases include EuPathDB or Ensembl. These databases contain genomic and transcriptomic data from model organisms as well as from various other eukaryotic lineages.
Hereafter we will be using GenBank.
Keyword searches on GenBank
The default GenBank page is a search page which allows us to search for sequences using various keywords.

In the left side of the search we can select the database which we want to search (e.g. nucleotide, protein, genome). Also, in the keyword searches, logical arguments like AND, OR, NOT can be used together various categories. The categories are mentioned in square brackets. e.g. [Organism].
Example:
Rodentia[Organism] AND 12S rRNA[Gene Name] NOT Mus[Organism]
When defining names for various organisms, please make sure the organisms queried are spelled exactly as in GenBank’s taxonomic database or use their respective unique taxon ID (for Rodentia it is txid9989[Organism]).
You can build your query using Advanced search. Use Help for advice how to construct it.
An advantage of this approach is that the result can be easily filtered to complete sequences, sequence type (mRNA, genomic, protein) or taxonomic tree.
Searches Using Sequence Homology (BLAST -- Basic Local Alignment Search Tool) on GenBank. https://blast.ncbi.nlm.nih.gov/Blast.cgi
A disadvantage of keyword search is that genes annotated differently from the query are not retrieved. Also, using keywords can be quite laborious, for example if you want to download sequences from non-model taxon representatives. In these cases we use homology searches, such as BLAST. Basic types of BLAST are:
BLASTN (Nucleotide BLAST) – the query is a nucleotide sequence, searched against a nucleotide database.
BLASTP (Protein BLAST) – the query is a protein sequence, searched against a protein database.
BLASTX – the query is a nucleotide sequence which will be translated in six frames, searched against a protein database (takes longer because it represents multiple searches per query).
TBLASTN – the query is a protein sequence, searched against a nucleotide database translated into protein space.
TBLASTX - the query is a nucleotide sequence which will be translated in six frames, searched against nucleotide databases translated into protein (takes longer).
Choose one of these approaches on the main page or change your choice using the tabs of the application page. Then enter your query sequence(s) into the large field.
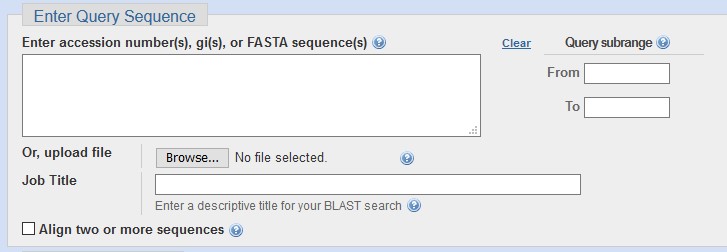
For BLASTX/TBLASTX, remember to select the correct Genetic code for your query.
In the section Choose Search Set, select a target database. If not selecting human or mouse, you can enter optional database parameters, such as taxon limit or other criteria.
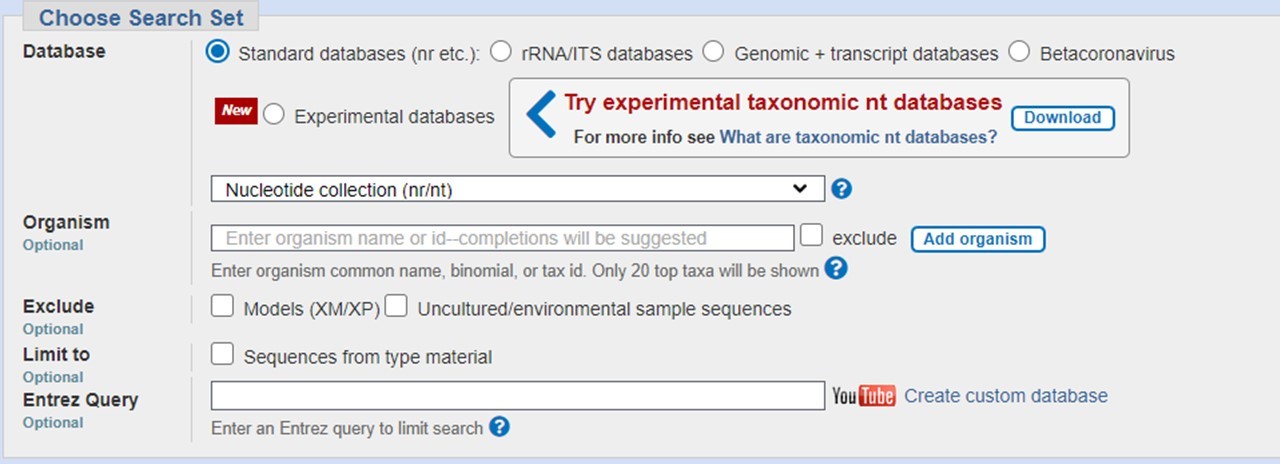
In the section Program Selection, choose a subtype of algorithm based on expected sequence similarity and start your search by clicking the “BLAST” button.

A disadvantage of this method is that sequences are retrieved based on similarity, not function. Therefore, a subsequent phylogenetic analysis is often helpful in resolving sequence homologies, for example within gene families.
Exercise 1
1. You have obtained a sequence for an unknown human gene, and you suspect it to encode a protein. We will try to find out which protein this is using BLASTX.
>Human-sequence
TGGCCAGGCTGGTGTCCAGATTGGCAATGCCTGCTGGGAGCTCTACTGCCTGGAACACGGCATCCAGCCCGATGGCCAGATGCCAAGTGACAAGACCATTGGGGGAGGAGATGACTCCTTCAACACCTTCTTCAGTGAGACGGGCGCTGGCAAGCACGTGCCCCGGGCTGTGTTTGTAGACTTGGAACCCACAGTCATTGATGAAGTTCGCACTGGCACCTACCGCCAGCTCTTCCACCCTGAGCAGCTCATCACAGGCAAGGAAGATGCTGCCAATAACTATGCCCGAGGGCACTACACCATTGGCAAGGAGATCATTGACCTTGTGTTGGACCG
Exercise 2
1. From the protist Paratrimastix pyriformis we obtained the following protein. We will look for homologous sequences in the database and assess its function by using the distantly related protein search tool PSI-BLAST (Position-Specific Iterated BLAST).
>Paratrimastix_pyriformis_protein
KSETLLERLRRRMQPTISCLSNGIRVVSLHDKNYVNDVRSIGIYFDVPQSNPDSLAPCHVLNQALSQGSSHLGAFPGTLSVQYSRDLISYRATFPREEKPDHALQLMANLWRRASSLTKSDTDAAAPFVSSRIEALRSDPQEVLNDLVHMRMFEGTALGLPLLHTRLEAGRCAELVPRDLDALLATPLAGQDRKWHAPPLKQLSNFVSSTHCPTRCVVVASGIPHDSLLQATKRHLTFATERIPSGSSGDRPCEKPGKFGAIHGHYISMPPDCFLDVDICYPTKGLADPHLVPLLLAEDAIGKASAFSAGGPGKGVLTRANMRSMGMPGVMRARAHMTTYGKQGFFGIHVGGDPAAARVFASVSAAEFMSGALEGFSDIEIRRAKSRLKASFSLAHEQQPYRMEDAARQVFARNLRGSGMETPKGASGSSGFLTVRGLADLIDRCPTQSVGQAMKELTCRCRPIVAWSHPESAFRFPEKGTSTPAPQIHRSNFGGWSDSHKGSPDICEYRPSPRNDRCHLEGPTSI
You can set the PSI-BLAST algorithm in the
Program Selectionsection of BLASTP.
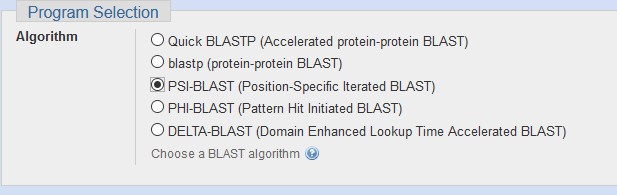
2. Use the sequence filter in the upper right to limit your next search, or simply click RUN to repeat the search using a position-specific score matrix (PSSM) derived from the top hits.
The first run of the PSI-BLAST is actually a normal BLASTP. The results of this search are used to calculates a profile or a position-specific score matrix (PSSM) from the multiple alignment. The PSSM captures the conservation pattern in alignment and stores it as a matrix of scores for each position in the alignment-highly conserved positions receive high scores and weakly conserved positions receive scores near zero. This profile is used in place of the original substitution matrix for a further search of the database to detect sequences that match the conservation pattern specified by the PSSM. This way you are able to retrieve more distant sequence homologs.
3. Iterate your search until you find satisfactory results or until the algorithm reaches convergence, i.e. it cannot find any new hits (marked yellow in the Descriptions field).
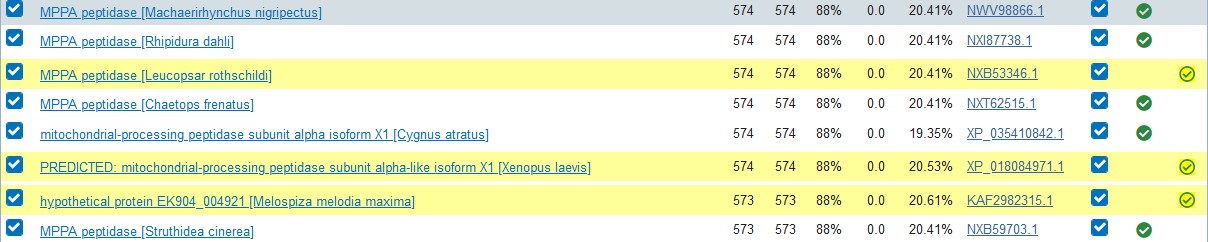
4. Access your recent results using the Recent Results link in the upper right part of the page.
An even more sensitive approach than PSI-BLAST is based on Hidden Markov Models. Online, this can be done on the HHpred webserver.
Sequence Formats
FASTA - plain-text, simple format, usually the input format for many sequence analysis programs.
>seqname1 description of sequence
AACTGCCATGCAACCATGACTAGCATA
GenBank - metadata-rich format, shown by default for sequences in GenBank, e.g. www.ncbi.nlm.nih.gov/nuccore/NC_001603.2.
Downloading sequences
Sequences from GenBank can be downloaded one by one or in batch.
- Downloading sequences one by one
- Batch downloading of the sequences
Let’s try this with a chloroplast-encoded gene from Euglena gracilis
In the top left corner of each sequence record there is a link called FASTA. Clicking on the link will show the entire sequence in FASTA format.
Using Copy Ctrl(Cmd) + C and Paste Ctrl(Cmd) + V, we can copy the sequence from the webpage and paste it in Notepad++ (gedit in Ubuntu, BBedit in MacOS). Afterwards we can save the file as .fasta.
Another way to save the sequence is the top right menu Send to:. From here we can choose to save the entire sequence or just the coding sequences, and we can select various file formats.
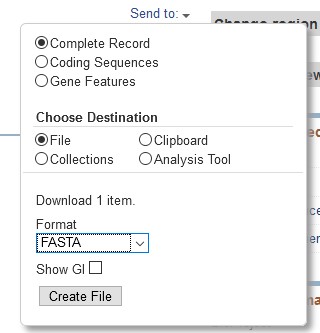
After selecting the format we click on Create File and we save the file.
Both keyword and sequence similarity search approaches allow you to download the results as sequence batches.
For the keyword approach: Select the sequences of interest using the checkbox on the left (or check none to download all). Using the Send to: button, define the parameters of the output.
For BLAST hits: On the result page, select the sequences of interest using the checkbox or using the Select all button. Clicking the Download starts the export process by letting you choose the output format.
Sequence editing
For basic editing of your sequence datafiles you can use freeware editors, e.g. UGENE/AliView.
We will mainly focus on AliView.
AliView basics:
Input formats – FASTA, Phylip, Nexus and others (not GenBank unfortunately)
Double-click a sequence name to view/edit it. Check sequence statistics using the graph tab on the right.
Ctrl(Cmd) + Ato select all.Toggle translation using a button (see if sequences are in frame):

There are many other features of AliView, however in many cases for advanced features, commercial software like Geneious is more suitable in these cases (ask for a license at the IT department if interested).
Exercise 3
1. Download all available enolase sequences from the phylum Parabasalidea. Use a sequence homology search of BLAST and retrieve one sequence per species. As query, use the Trichomonas vaginalis enolase sequence:
>gi|123479346|ref|XM_001322797.1| Trichomonas vaginalis G3 enolase, putative (TVAG_282090) mRNA, complete cds
ATGGAAGCAACAGCTGAAGTTACAGAGGAACCACAAGGAGATACTCCAGAAATCGTAGAAGCAAAGAGAAAATATCTTGTTGACTACGATATTCAAAATGTTCTTCATGAAGCTCTCAATGAATTAATGGAATCACGTCCTGAAGATGTTTTTGGAGTTTTATCACGAATTTTTGAACGTCGCGCTGCAAAACCAATTATTGATCATGTTTTAGCTCGAGAAGTCCTTGATTCAAGAGGTAATCCCACAGTTGAAGTTGACGTTTATGCAAAATACTTAAATACCGTAGAATTTGTTGCTAGATCTTCTTCTCCATCAGGAGCATCAACAGGATCAAAGGAAGCAAAAGAACTCAGAGATGGAGATAATAGATTCGGTGGTAAGGGCGTTACACATGCAGTCAAGAACGTCAATACAATTATTAGCAAAGCAATTGCAGGCAAATTACTCGAAAATCTTGCAGAAATTGATAACGCAATTATTGCAGCCGATGGAACAGAACTCAAGGAAAAACTTGGCGGAAATGCAACCACAGCAACTTCTTTTGCCGTTGCTACAGCAGGGGCAGCTATTAGACATGAAGAATTATTTATTTACCTTGCCAGACAATTCCATGAAGAAATGCCAAAGAAATTTAAGCTTCCAGCACTTTTCTTCAATATATTAAACGGTGGAAAGCACGCTGGTGGAAATTTGAAGATCCAAGAATTTATGATTTCTCCACGAACAGATATCAGCTTTCCTGAGCAGCTAAGAATGATTGGAGAAATATACCAGAAACTTGGCCAGGTTGTTGTCAAGAAATATGGCGTTTCTGCAAAGAATCTTGGCGATGAAGGTGGCTATGCACCAGCTCTTAACACTCCCGAGGAAGCTCTCGAAGTAATTGAAAGAGCAGCAAATCTTTGCGGCTACCAACCAGGCAGCGATGTCTTCTTTGCACTAGATGCAGCTGCATCTGAATTTTATGATGCAACAAAGAAGCAATACGAAATTCTTCCAAACGTATGGAAGACTGGTGATGAAATGATTGAGTTTTGGAAAGATTTGATCGCTAAACATCCAGCAATTATTTCAATTGAAGACGGACTTGAAGAAAAAGACTACGAAACATGGATTAAACTCAATGAGCAGCTCGGTAGCAAGATACAGTTGGTAGGAGATGATCTTTACACAACAAATCCAAAGATGATTGAGCAGGGAATCGAAAAGAAGTGGTGCAACGCATTGTTGATGAAAGTAAACCAAATAGGAACAATTACAGAGGCAATGAAAGCTGCAAAACTCGTTCTCTCTGCAGGCCAGAAAGTCATGGTTTCTCACAGATCAGGAGAAACATGTAACTCTTTAATTTCTGACTTAGCTGTTGCAATTGGAGCTCAGTCAATTAAAGCTGGCTCTTGCGCAAGAGGAGAGAGAATACAGAAATACACAAGATTGCTACAGATCTATGAATACTTGAGAGATAATAATATGCTTTAA
2. Do a keyword search for cytochrome b[protein name] from the broadest possible selection of primates (one sequence per species but 30 sequences at most). To avoid partial sequences limit the search to sequnec lengths between amino acids - 300:400 [slen]. Make sure the file can be imported to AliView.
BASH command line basics
For this make sure you installed the linux subsystem for Windows, based on the instructions which you got by e-mail. For MacOS or Linux users, there is a usable terminal already.
Basic commands which you will use in the practicals. Mac and Linux users, open the Terminal. For Windows users, click on start and start typing Ubuntu. Click on the Ubuntu app, and a new terminal window will appear.
To see your current working directory you can use the command:
pwd
#### pwd stands for Print Working Directory
To make a new directory and change to it you can use the commands:
mkdir moltax
cd moltax
mkdir stands for MaKe DIRectory
cd stands for Change Directory
You can copy and paste into your terminal using right-click (Windows subsystem), Ctrl+Shift+V (Ubuntu) or CMD+V (MacOS).
Writing "cd" without specifying a directory will navigate you back to your home directory.
Windows users only!. ---> In the subsystem, you can use explorer.exe . to open the current working directory in explorer. This will be useful when you will need to visualize tress which you computed on the subsystem. The dot . after explorer.exe is critical, otherwise the command will not work.
Similarly, writing open . in a terminal of a Mac, or nautilus . in Ubuntu will do the same thing.
Into the open window, you can now drag and drop files from your system. You can also copy your results from the linux environment to other locations. So let's do that with your FASTA files.
To list the files in the current directory:
ls -la
#### ls stands for LiSt, -l and -a are parameters to show the long tabular format of all files, including hiddens
To quickly look into the content of a file:
less FILE
#### throughout the course, replace anything marked as PLACEHOLDERS with an actual file or path
To make a CoPy or rename/MoVe a file in the terminal:
cp PATH/TO/FILE PATH/TO/COPY
mv PATH/TO/FILE PATH/TO/NEWFILE