Part 7: Metagenomics
Part 7: Metagenomics
Metagenomics is a broad term, which includes a large number of approaches to obtain sequencing data from various samples and their subsequent processing.
The two main approaches are:
1) Amplicon sequencing - involves sequencing parts of the 16S/18S rRNA from the environment.
2) Shotgun metagenomics - which involves sequencing of the entire genetic material obtained from the environment.
Amplicon sequencing is used to get taxonomic information on the composition of a community of microorganisms and to some extent we can quantify the number of individual species. The second approach also provides functional information about the community, but at a significantly higher sequencing cost.
There is also a third emerging approach, called metatranscriptomics, where the total RNA isolated from environmental samples is sequenced. This method provides also functional information, however, it is really challenging to define which organism from the sample performs which function.
Next, we will focus only on amplicon sequencing.
The most used programs for processing amplicon data include, for example, QIIME or Mothur.
Geneious also can be used to partially process amplicon data. Instructions can be found here.
Exercise 1
The exercise will be performed on Galaxy server. We will use the following tutorial.
Exercise 2
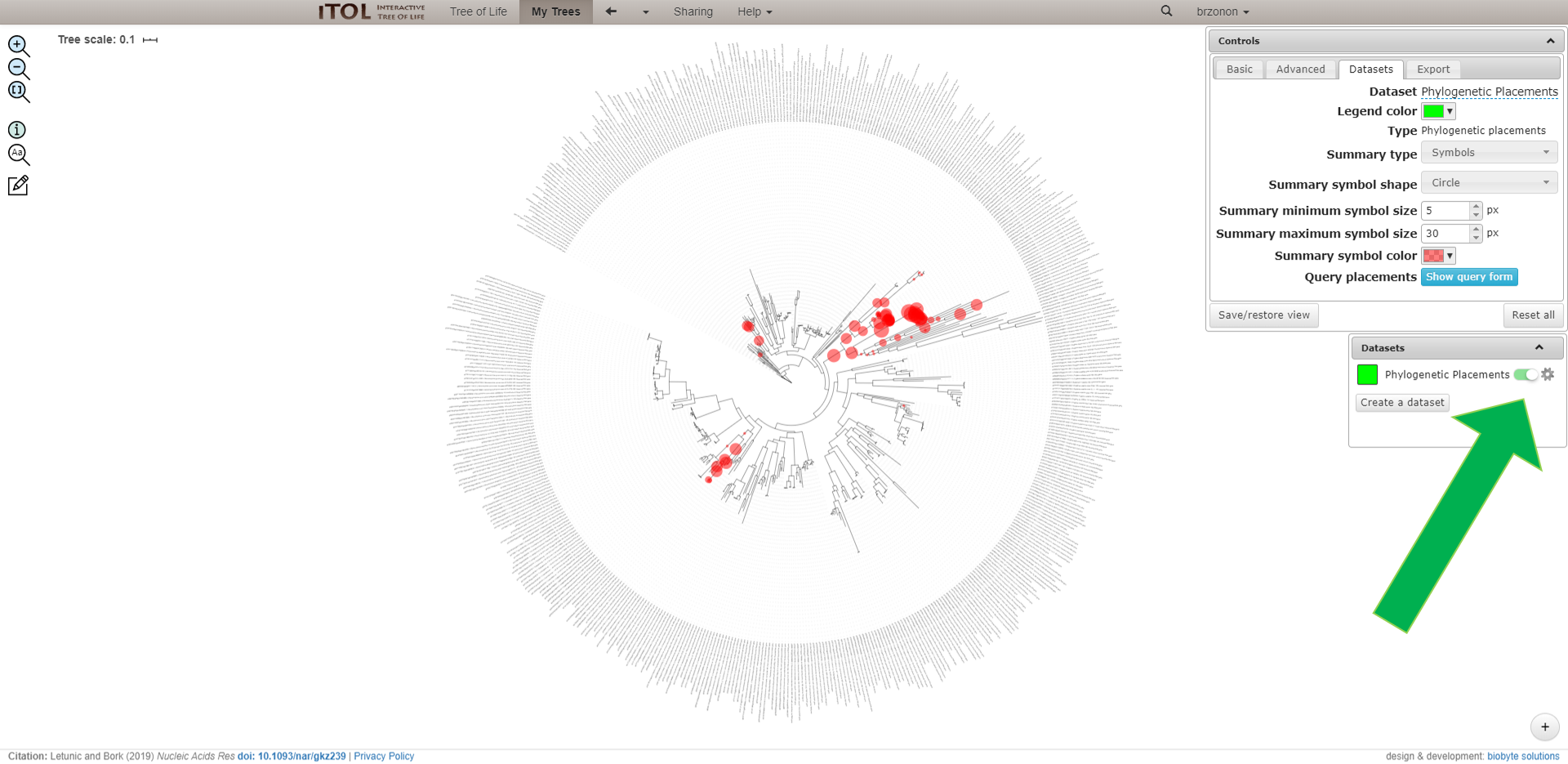
Adding amplicon data on a reference tree.
In this exercise we will try to add our amplicon sequences to a reference tree which was already computed.1) Download the reference tree from here
2) We align the amplicon sequences to our reference alignment using MAFFT --add function.
3) We trim the alignment using trimal with the following parameters:
trimal -in infile -out outfile -gt 0.01
This is a very relaxed trimming, as we align very short sequences to a full-length 18S alignment. For this reason, we use -gt 0.01 meaning that we want to remove only columns in which more than 99% of the sequences have gaps.
4) We add the sequences to the reference tree using EPA RaxML (The Evolutionary Placement Algorithm, Randomized Axelerated Maximum Likelihood) with -f v parameter.
raxmlHPC-PTHREADS-SSE3 -f v -G 0.2 -m GTRCATI -n EPARUN -s trimmed_alignment -t reference_tree -T 4
5) The computed tree can be visualized using iTOL